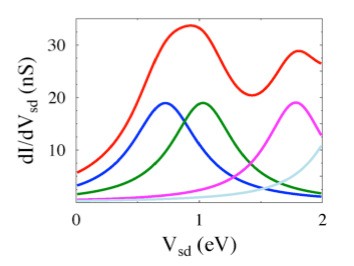
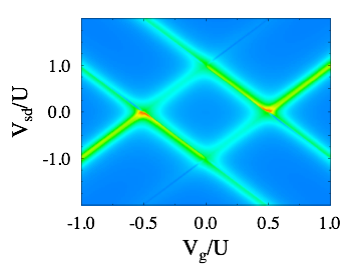
Quantum transport in molecular states language
Most ab initio simulations of molecular transport junctions employ language of effective single-particle orbitals. This approach becomes inadequate at resonance due to sensitivity of molecules to oxidation/reduction/excitation. We develop theoretical methods capable of describing transport in the language of molecular many-body states. In particular, we consider generalization of two popular approaches to quantum transport formulated: quantum master equation (QME) and non-equilibrium Green functions (NEGF). When developed the schemes will allow to incorporate equilibrium quantum chemistry methods into description of open non-equilibrium molecular systems. It will also provide a convenient setup to treat non-adiabatic dynamics in molecular junctions.